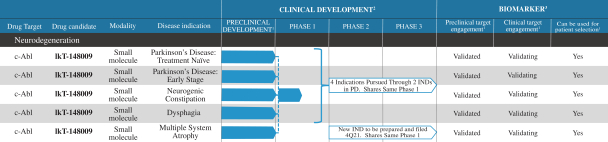
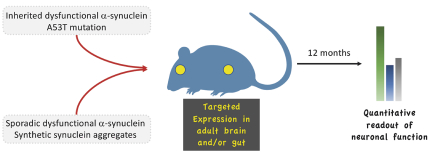
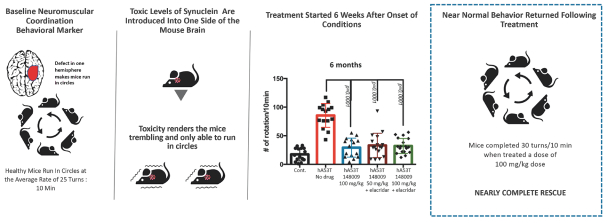
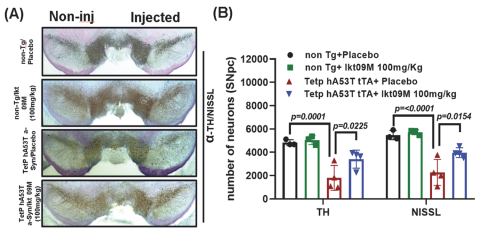
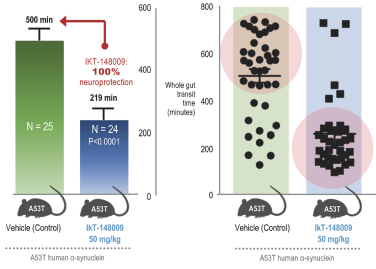
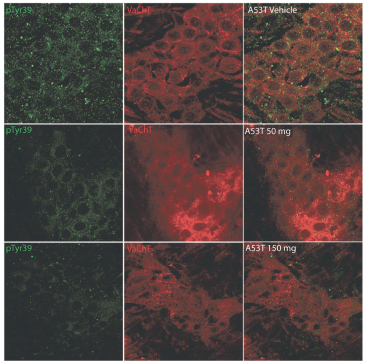
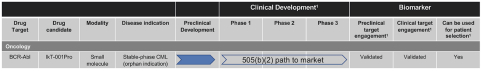
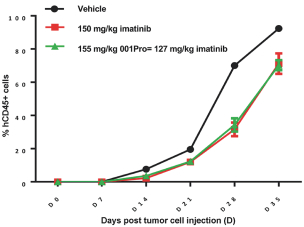
Filed Pursuant to Rule 424(b)(4)
Registration No. 333-257032
PROSPECTUS
15,000,000 Shares
Common Stock

Inhibikase Therapeutics, Inc.
This is a firm commitment public offering of 15,000,000 shares of common stock, par value $0.001 per share, of Inhibikase Therapeutics, Inc. The public offering price of our shares is $3.00 per share.
Our common stock is listed on the Nasdaq Capital Market (“Nasdaq”) under the symbol “IKT.” On June 15, 2021, the last reported sale price of our common stock was $4.04 per share.
We are an “emerging growth company” and a “smaller reporting company,” each as defined under the federal securities laws and, as such, have elected to comply with certain reduced reporting requirements for this prospectus and may elect to do so in future filings. See the section titled “Implications of Being an Emerging Growth Company and a Smaller Reporting Company.”
Investing in our securities involves a high degree of risk. See “Risk Factors” beginning on page 15 of this prospectus and elsewhere in this prospectus for a discussion of information that should be considered in connection with an investment in our securities.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense. The securities are not being offered in any jurisdiction where the offer is not permitted.
| Per Share |
Total(1) | |||||||
| Public offering price |
$ | 3.00 | $ | 45,000,000 | ||||
| Underwriting discounts and commissions(1) |
$ | 0.21 | $ | 3,150,000 | ||||
| Proceeds to us, before expenses |
$ | 2.79 | $ | 41,850,000 | ||||
| (1) | Assumes no exercise of the underwriters’ option to purchase additional shares of our common stock as described below. The registration statement, of which this prospectus is a part, also registers for sale warrants to purchase 750,000 shares of our common stock to be issued to the representative of the underwriters. We have agreed to issue the warrants to the representative of the underwriters as a portion of the underwriting compensation payable to the underwriters in connection with this offering. See “Underwriting” beginning on page 178 for a description of compensation payable to the underwriters. |
We have granted a 45-day option to the representative of the underwriters to purchase up to 2,250,000 additional shares of common stock from us solely to cover over-allotments, if any, at the public offering price, less underwriting discounts and commissions.
The underwriters expect to deliver the shares of common stock to purchasers on or about June 18, 2021, subject to customary closing conditions.
ThinkEquity
a division of Fordham Financial Management, Inc.
JonesTrading
The date of this prospectus is June 15, 2021.